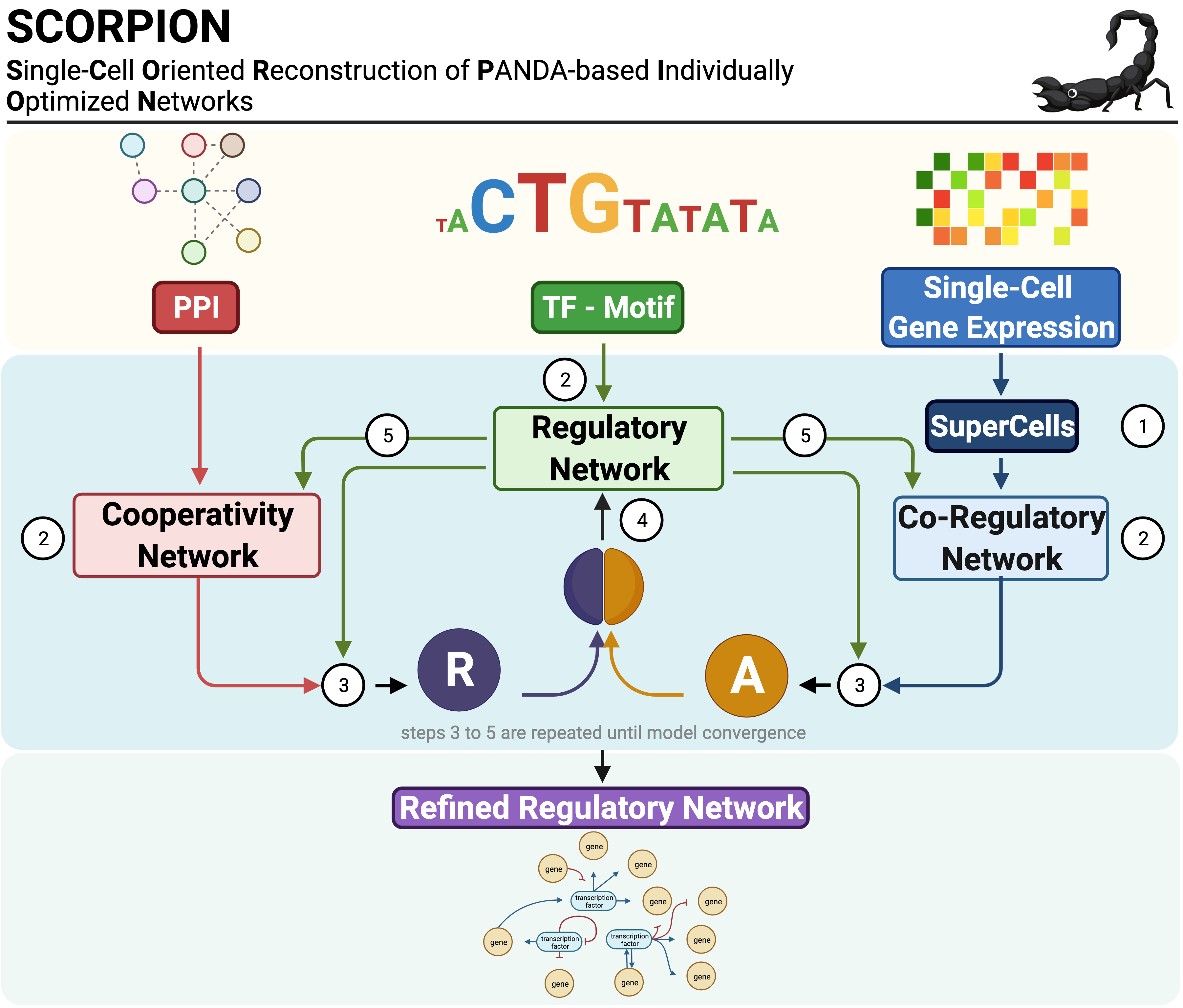
SCORPION (Single-Cell Oriented Reconstruction of PANDA Individually Optimized Gene Regulatory Networks) is a Python package for constructing gene regulatory networks from single-cell and single-nucleus RNA sequencing data. The package addresses the sparsity inherent in single-cell expression data through coarse-graining, which aggregates similar cells to improve correlation structure detection. Network reconstruction is performed using the PANDA (Passing Attributes between Networks for Data Assimilation) message-passing algorithm, integrating transcription factor motifs, protein-protein interactions, and gene expression data. By using consistent baseline priors across samples, SCORPION produces comparable, fully-connected, weighted regulatory networks suitable for population-level analyses.
pip install pyscorpionTo install the development version from GitHub:
pip install git+https://github.com/kuijjerlab/pySCORPION.gitfrom scorpion import load_example_data, scorpion, run_scorpion
# Load the bundled example dataset
data = load_example_data()
# Construct a single regulatory network
network = scorpion(
gex_matrix=data["gex_matrix"],
tf_motifs=data["tf_motifs"],
ppi_net=data["ppi_net"],
)
# Construct networks stratified by cell groups
networks = run_scorpion(
gex_matrix=data["gex_matrix"],
tf_motifs=data["tf_motifs"],
ppi_net=data["ppi_net"],
cells_metadata=data["metadata"],
group_by="region",
)The package ships with a colorectal cancer single-cell RNA-seq dataset (300 genes, ~1 950 cells, 3 donors, 3 tissue regions) that can be loaded with a single call:
from scorpion import load_example_data
data = load_example_data()
data["gex_matrix"] # Gene expression matrix (genes × cells)
data["tf_motifs"] # TF–target motif prior network
data["ppi_net"] # Protein–protein interaction network
data["metadata"] # Cell-level metadata (cell_id, donor, region, cell_type)Constructs a single gene regulatory network from a gene expression matrix using coarse-graining and the PANDA algorithm.
Usage:
result = scorpion(
gex_matrix=expression_data,
tf_motifs=None,
ppi_net=None,
computing_engine="cpu",
n_cores=1,
gamma_value=10,
n_pc=25,
assoc_method="pearson",
alpha_value=0.1,
hamming_value=0.001,
n_iter=float("inf"),
out_net=["regNet", "coregNet", "coopNet"],
z_scaling=True,
show_progress=True,
randomization_method=None,
scale_by_present=False,
filter_expr=False,
random_state=None,
gene_names=None,
)Parameters:
| Parameter | Description | Default |
|---|---|---|
gex_matrix |
Expression matrix with genes in rows and cells in columns | Required |
tf_motifs |
DataFrame with columns [TF, target gene, motif score]. Pass None for co-expression analysis only |
None |
ppi_net |
DataFrame with columns [protein1, protein2, interaction score]. Pass None to disable PPI integration |
None |
computing_engine |
Computation backend: "cpu" or "gpu" |
"cpu" |
n_cores |
Number of processors for BLAS/MPI parallel computation | 1 |
gamma_value |
Coarse-graining level; ratio of cells to super-cells | 10 |
n_pc |
Number of principal components for kNN network construction | 25 |
assoc_method |
Gene association method: "pearson", "spearman", or "pcNet" |
"pearson" |
alpha_value |
Weight of prior networks relative to expression data (0–1) | 0.1 |
hamming_value |
Convergence threshold based on Hamming distance | 0.001 |
n_iter |
Maximum number of PANDA iterations before stopping | inf |
out_net |
Networks to return: "regNet", "coregNet", and/or "coopNet" |
All three |
z_scaling |
Return Z-score normalized edge weights; False returns [0,1] scale |
True |
show_progress |
Print progress messages during computation | True |
randomization_method |
Randomization for null models: None, "within_gene", or "by_gene" |
None |
scale_by_present |
Scale correlations by percentage of cells with non-zero expression | False |
filter_expr |
Remove genes with zero expression across all cells before inference | False |
random_state |
Random seed for reproducibility | None |
gene_names |
Array of gene names; if None, inferred from the expression matrix |
None |
Return value:
A dictionary containing:
| Component | Description |
|---|---|
regNet |
Regulatory network matrix (TFs × target genes) |
coregNet |
Co-regulation network matrix (genes × genes) |
coopNet |
TF cooperative network matrix (TFs × TFs) |
numGenes |
Number of genes in the network |
numTFs |
Number of transcription factors |
numEdges |
Total number of edges in the regulatory network |
Constructs regulatory networks for multiple cell groups defined by metadata columns. This function wraps scorpion() to enable stratified network inference and returns results in a format suitable for comparative analysis.
Usage:
networks = run_scorpion(
gex_matrix=expression_data,
tf_motifs=motif_prior,
ppi_net=ppi_network,
cells_metadata=metadata,
group_by="region",
normalize_data=True,
remove_batch_effect=False,
batch=None,
min_cells=30,
)Parameters:
| Parameter | Description | Default |
|---|---|---|
gex_matrix |
Expression matrix with genes in rows and cells in columns | Required |
tf_motifs |
DataFrame with columns [TF, target gene, motif score] | Required |
ppi_net |
DataFrame with columns [protein1, protein2, interaction score] | Required |
cells_metadata |
DataFrame with cell-level metadata; must contain columns specified in group_by |
Required |
group_by |
String or list of column name(s) in cells_metadata for stratification |
Required |
normalize_data |
Apply log normalization to expression data before network inference | True |
remove_batch_effect |
Perform batch effect correction before network inference | False |
batch |
Column name in cells_metadata giving batch assignment for each cell; required if remove_batch_effect=True |
None |
min_cells |
Minimum number of cells required per group to build a network | 30 |
Additional parameters: All scorpion() parameters (computing_engine, n_cores, gamma_value, n_pc, assoc_method, alpha_value, hamming_value, n_iter, out_net, z_scaling, show_progress, randomization_method, scale_by_present, filter_expr) can be passed to control network inference behavior. See scorpion() documentation above.
Return value:
A DataFrame in wide format where:
- Rows represent TF-target pairs
- Columns represent network identifiers (derived from
group_byvalues) - Values are edge weights from each network
Example output:
| tf | target | P31--T | P31--B | P31--N | P32--T | ... |
|---|---|---|---|---|---|---|
| AATF | ACKR1 | -0.326 | -0.337 | -0.344 | -0.298 | ... |
| ABL1 | ACKR1 | -0.340 | -0.339 | -0.351 | -0.312 | ... |
Examples:
from scorpion import load_example_data, scorpion, run_scorpion
# Load example data
data = load_example_data()
# Stratify by tissue region
nets_by_region = run_scorpion(
gex_matrix=data["gex_matrix"],
tf_motifs=data["tf_motifs"],
ppi_net=data["ppi_net"],
cells_metadata=data["metadata"],
group_by="region",
)
# Stratify by multiple variables
nets_by_donor_region = run_scorpion(
gex_matrix=data["gex_matrix"],
tf_motifs=data["tf_motifs"],
ppi_net=data["ppi_net"],
cells_metadata=data["metadata"],
group_by=["donor", "region"],
)
# With batch effect correction
nets_corrected = run_scorpion(
gex_matrix=data["gex_matrix"],
tf_motifs=data["tf_motifs"],
ppi_net=data["ppi_net"],
cells_metadata=data["metadata"],
group_by="region",
remove_batch_effect=True,
batch="donor",
)Performs statistical testing on network edges to identify differential regulatory relationships between groups. The function supports single-sample tests, two-sample comparisons, and paired tests. All computations are fully vectorized for efficiency with large-scale datasets.
Usage:
results = test_edges(
networks_df=nets,
test_type="single",
group1=tumor_nets,
group2=None,
paired=False,
alternative="two.sided",
padjust_method="BH",
min_log2fc=0.0,
moderate_variance=True,
empirical_null=True,
)Parameters:
| Parameter | Description | Default |
|---|---|---|
networks_df |
Output from run_scorpion() |
Required |
test_type |
Test type: "single" (one-sample) or "two.sample" |
Required |
group1 |
List of column names for the first (or only) group | Required |
group2 |
List of column names for the second group (two-sample tests) | None |
paired |
Perform paired t-test; requires equal-length groups in matched order | False |
alternative |
Alternative hypothesis: "two.sided", "greater", or "less" |
"two.sided" |
padjust_method |
Multiple testing correction method (see statsmodels.stats.multitest) |
"BH" |
min_log2fc |
Minimum absolute log2 fold change for inclusion (two-sample/paired only) | 0.0 |
moderate_variance |
Apply SAM-style variance moderation; adds median(SE) to denominator | True |
empirical_null |
Use Efron's empirical null (median/MAD) for p-value calibration | True |
Return value:
A DataFrame containing:
| Column | Description |
|---|---|
tf, target |
TF-target pair identifiers |
meanEdge |
Mean edge weight (single-sample) |
meanGroup1, meanGroup2 |
Group means (two-sample) |
diffMean |
Difference in means, Group1 − Group2 (two-sample) |
cohensD |
Cohen's d effect size (two-sample and paired tests) |
log2FoldChange |
Log2 fold change (two-sample) |
tStatistic |
t-statistic |
pValue |
Raw p-value |
pAdj |
Adjusted p-value |
Examples:
from scorpion import test_edges
# Define groups
tumor_nets = [c for c in nets_by_donor_region.columns if c.endswith("--T")]
normal_nets = [c for c in nets_by_donor_region.columns if c.endswith("--N")]
# Two-sample comparison: Tumor vs Normal
results = test_edges(
networks_df=nets_by_donor_region,
test_type="two.sample",
group1=tumor_nets,
group2=normal_nets,
)
# Paired test for matched samples (same patient)
tumor_ordered = ["P31--T", "P32--T", "P33--T"]
normal_ordered = ["P31--N", "P32--N", "P33--N"]
results_paired = test_edges(
networks_df=nets_by_donor_region,
test_type="two.sample",
group1=tumor_ordered,
group2=normal_ordered,
paired=True,
)
# Single-sample test: edges differing from zero
results_single = test_edges(
networks_df=nets_by_region,
test_type="single",
group1=tumor_nets,
)Performs linear regression to identify edges with significant trends across ordered conditions. This is useful for studying disease progression or developmental trajectories.
Usage:
results = regress_edges(
networks_df=nets,
ordered_groups=ordered_conditions,
padjust_method="BH",
min_mean_edge=0.0,
)Parameters:
| Parameter | Description | Default |
|---|---|---|
networks_df |
Output from run_scorpion() |
Required |
ordered_groups |
Dictionary where each value is a list of column names in networks_df. Keys represent ordered conditions (e.g., {"Normal": [...], "Border": [...], "Tumor": [...]}). The order of keys defines the progression |
Required |
padjust_method |
Multiple testing correction method | "BH" |
min_mean_edge |
Minimum mean absolute edge weight for inclusion | 0.0 |
Return value:
A DataFrame containing:
| Column | Description |
|---|---|
tf, target |
TF-target pair identifiers |
slope |
Regression slope (change per condition step) |
intercept |
Regression intercept |
rSquared |
Coefficient of determination |
fStatistic |
F-statistic for the regression |
pValue |
Raw p-value |
pAdj |
Adjusted p-value |
meanEdge |
Overall mean edge weight |
mean<Condition> |
Mean edge weight for each condition |
Example:
from scorpion import regress_edges
# Define ordered progression: Normal → Border → Tumor
normal_nets = [c for c in nets_by_donor_region.columns if c.endswith("--N")]
border_nets = [c for c in nets_by_donor_region.columns if c.endswith("--B")]
tumor_nets = [c for c in nets_by_donor_region.columns if c.endswith("--T")]
ordered_conditions = {
"Normal": normal_nets,
"Border": border_nets,
"Tumor": tumor_nets,
}
# Identify edges with significant trends
results_reg = regress_edges(
networks_df=nets_by_donor_region,
ordered_groups=ordered_conditions,
)
# Edges increasing along progression
increasing = results_reg[(results_reg["pAdj"] < 0.05) & (results_reg["slope"] > 0)]
# Edges decreasing along progression
decreasing = results_reg[(results_reg["pAdj"] < 0.05) & (results_reg["slope"] < 0)]- NumPy (>=1.21)
- pandas (>=1.3)
- SciPy (>=1.7)
- scikit-learn (>=1.0)
- igraph (>=0.10)
If you use SCORPION in your research, please cite:
Osorio, D., Capasso, A., Eckhardt, S.G. et al. Population-level comparisons of gene regulatory networks modeled on high-throughput single-cell transcriptomics data. Nature Computational Science 4, 237–250 (2024). https://doi.org/10.1038/s43588-024-00597-5
- Supplementary Materials: https://github.com/dosorio/SCORPION/
- Issue Tracker: https://github.com/dcosorioh/pySCORPION/issues
- R Package (CRAN): https://cran.r-project.org/package=SCORPION