{kind=link}
This is a general NER toolkit for BERT model training, evaluation, and prediction. The BIO_pipeline processes annotated corpora (with labels) and creates word-token level documents (.io or .bio) for training and evaluation. A split of development set and evaluation set should be considered. The Training_pipeline inputs the word-tokens (with labels) from development set, and train a BERT-familiy model. Checkpoints are saved to disk. The Evaluation_pipeline use a specified checkpoint to predict on the evaluation set, and perform entity-level evaluation. Both exact matching and partial matching with precision, recall, and F1 are reported. Once a final/ production NLP model is made and saved to disk, the Prediction_pipeline use it to predict.
Framework: PyTorch, Transformers
Compatible annotation tools: Label-studio, MAE, BRAT

Prepare project file system. We create folders as below (all modules under "pipelines" can be cloned from this repository):
Project folder
- BIO
- train_test_id
- pipelines
- BIO_pipeline.py
- Training_pipeline.py
- Evaluation_pipeline.py
- Prediction_pipeline.py
- configs
- configs_run_0.yaml
- modules
- __init__.py
- Prediction_utilities.py
- Training_utilities.py
- Utilities.py
Edit config file. All the pipelines use the same config file.
The parameters for word-token (IO/BIO) creation are:
########################
# BIO processing parameters
########################
txt_dir: ~\ADE medication NER\data\text
ann_dir: ~\ADE medication NER\data\ann
BIO_dir: ~\ADE medication NER\BIO
BIO_mode: BIO
The output from BRAT annotation tool is .ann format. If different annotation tool was used, modify the children class in BIO_pipeline. The BIO_dir specifies the output directory for .io or .bio files. BIO_mode specifies whether the labels will include "B-" and "I-" (BIO) or without (IO). When 1) training data is sufficient or 2) same type of entities could show up back-to-back, BIO is suggested. Otherwise IO could be more efficient (reduces the categories for prediction by half). Once executed, the BIO_pipeline creates .io or .bio files in BIO_dir.
The parameters for NLP model fine-tuning are:
########################
# Model fine-tune parameters
########################
# development set file path
deve_id_file: [deve_id_path:str]
# ratio of validation set. Will be sampled from development set
valid_ratio: [ratio:float]
# Define entity labels and category numbers
label_map:
O: 0
label_1: 1
label_2: 2
# tokenizer path
tokenizer: [tokenizer:str]
# Word tokens to be included in a segment
word_token_length: [length:int]
# step of sliding window for training instance creation
slide_steps: [steps:int]
# wordpiece tokens to include in a training instance
wordpiece_token_length: [length:int]
# base NLP model file path
base_model: [model_path:str]
# learning rate
lr: [rate:float]
# n_ephoch
n_epochs: [N:int]
# batch_size
batch_size: [size:int]
# Output path
out_path: [path:str]
deve_id_file specifies the id list (per document_id per row) for development set. The pipeline will use the ids to pull .io or .bio files. Training and validation split is handled automatically, just specify the ratio in valid_ratio. label_map defines the entity types and a numeric code. "O" means other (non-entity). word_token_length specifies the number of word-tokens to include in a training instance. slide_steps speficies the number of word-tokens to slide to create the next training instance. wordpiece_token_length defines the exact number of tokens input into BERT. Padding and truncating applies. out_path specifies the root folder for checkpoints and logs. The Training_pipeline creates the following files:
out_path
- checkpoints
- [run name 1]
- checkpoints 1
- checkpoints 2
...
- [run name 2]
...
- logs
- [run name 1]
- log
...
The parameters for NLP model evaluation are:
########################
# Evaluation parameters
########################
# test set file path
test_id_file: [eval_id_path:str]
# checkpoint path
checkpoint_dir: [checkpoint path:str]
# checkpoint filename to evaluate, or "best" to pull the checkpoint with min validation loss.
checkpoint: [checkpoint file:str]
test_id_file specifies a list of document_id in evaluation set. The pipeline will pull them from BIO_dir. checkpoint_dir specifies the directory with checkpoints. checkpoint could be the filename of checkpoint, or "best".
Once the config file is completed, run:
$cd [Project folder]
$python BIO_pipeline.py -c ./configs/configs_run_0.yaml
$python Training_pipeline.py -c ./configs/configs_run_0.yaml
$python Evaluation_pipeline.py -c ./configs/configs_run_0.yaml
The Prediction_pipeline provides a coding template for production. The input should be a dictionary:
{[document_id 0] : [text 0],
[document_id 1] : [text 1],
[document_id 2] : [text 2]
...}
The parameters in config are:
########################
# Prediction parameters
########################
# directory with documents to predict
predict_doc_dir: ~\ADE medication NER\data\text
# predict model
predict_model: ~\ADE medication NER\Production Model
# Output file
predict_outfile: ~\ADE medication NER\prediction.pickle
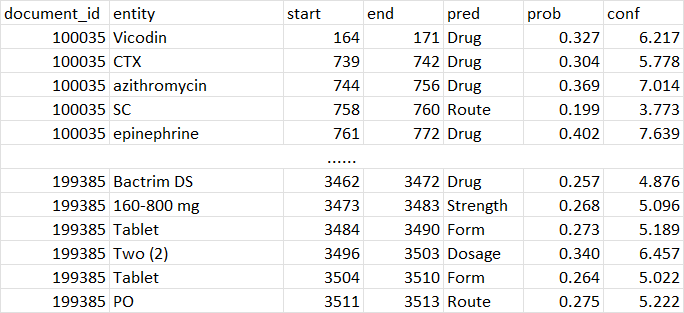
The output is a DataFrame with document_id, entity, start, end, pred, prob, conf. pred is the predicted entity type, prob is the average probability of all tokens in the entity, conf is the prob/baseline probability. Where baseline probability is the probability of randomly guess an entity type (1/N_entity_types).
We demo with 2018 i2b2 Adverse Drug Events & Medication Extraction (https://n2c2.dbmi.hms.harvard.edu/2018-challenge) datasets. The dataset include 505 annotated notes (303 training, 202 testing):
| Entity type | Total | Training set | Test set |
|---|---|---|---|
| Drug | 26,800 | 16,225 | 10,575 |
| Strength | 10,921 | 6,691 | 4,230 |
| Form | 11,010 | 6,651 | 4,359 |
| Dosage | 6,902 | 4,221 | 2,681 |
| Frequency | 10,293 | 6,281 | 4,012 |
| Route | 8,989 | 5,476 | 3,513 |
| Duration | 970 | 592 | 378 |
| Reason | 6,400 | 3,855 | 2,545 |
| ADE | 1,584 | 959 | 625 |
| Total | 83,869 | 50,951 | 32,918 |
We downloaded the dataset and distributed into the following folders:
ADE medication NER
- data
- ann
- 100035.ann
- 100039.ann
...
- text
- 100035.txt
- 100039.txt
...
We use BIO_pipeline to create BIO files:
$python BIO_pipeline.py -c ./configs/configs_i2b2.yaml
Now we have word-token files (with labels) in BIO folder:
ADE medication NER
- BIO
- 100035.bio
- 100039.bio
We use scripts/train test split.py to create train_id and test_id. Now we are ready to train models. Run Training_pipeline:
$python Training_pipeline.py -c ./configs/configs_i2b2.yaml
The checkpoint folder and log folder were created.
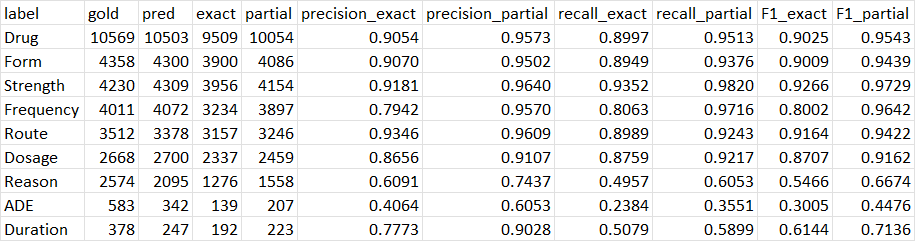
We then run Evaluation_pipeline. An evlauation metrics is created.
$python Evaluation_pipeline.py -c ./configs/configs_i2b2.yaml
Once we have a model, we can save if with scripts/save final model.py, and run Prediction_pipeline:
$python Prediction_pipeline.py -c ./configs/configs_i2b2.yaml
The predicted entities are saved to the outfile directory: